Definición y descripción
Malformación cerebral ocasionada por la incompleta división de los hemisferios cerebrales. Puede causar defectos en el desarrollo de la cara y en la estructura y el funcionamiento del cerebro. Se clasifican según la severidad:
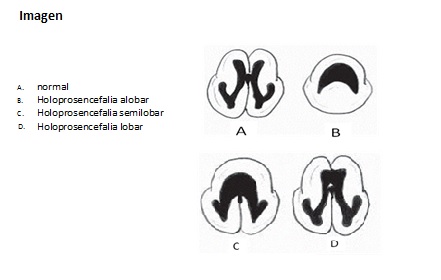
– Holoprosencefalia alobar: es la más grave, en la cual el cerebro no logra separarse, tiene un único ventrículo y se asocia generalmente a anomalías faciales severas, entre ellas la ciclopía.
– Holoprosencefalia semilobar: se caracteriza por tener separación en los hemisferios occipitales, pero fusión en los parietales y frontales.
– Holoprosencefalia lobar: en la cual existe una evidencia considerable de separación de los hemisferios del cerebro, permanece fusionada por lo general la zona más ventral y anterior de los hemisferios frontales.
Hay también variantes intermedias de estos casos.
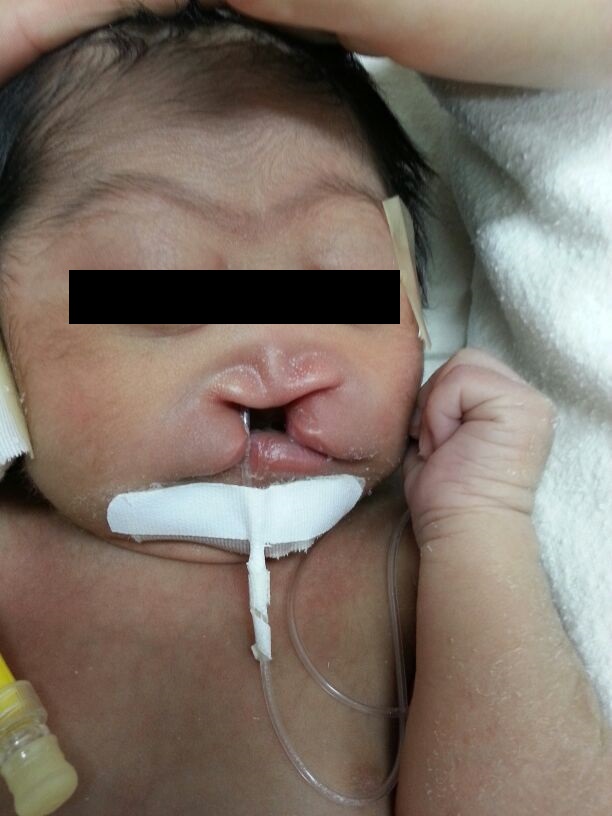
Se debe describir el tipo de holoprosencefalia, así como la presencia de anomalías faciales asociadas (ciclopía, hipotelorismo, proboscide, fisuras labiales de la línea media con agenesia de premaxila, etc.).
La holoprosencefalia puede asociarse también al Complejo Agnatia-otocefalia.
Etiología e impacto
Las formas alobares y semilobares tienen alta mortalidad durante los primeros meses de vida. Desde el punto de vista etiológico, la holoprosencefalia es heterogénea. Entre el 25-50% de todos los casos de holoprosencefalia se deben a causas cromosómicas numéricas o estructurales (Ej.: El síndrome de Patau por Trisomía 13). También existen síndromes génicos asociados a esta anomalía (entidades autosómicas dominantes y autosómicas recesivas)
El teratógeno mas frecuentemente asociado es la diabetes materna no controlada.
Aspecto genético del manejo inicial
El diagnóstico de la malformación se confirma con la neuroimagen. Como además de casos aislados hay casos sindrómicos, es importante acompañar la evaluación de otros órganos internos (ej.: riñón, corazón, etc.), así como evaluación de la función hipofisaria. Se sugiere realizar cariotipo en todos los casos y evaluación familiar (puede haber familiares afectados con formas mínimas, como un incisivo central único); buscar antecedente materno de diabetes, antecedentes de consanguinidad, etc.
Figura 2.1.8. Holoprosencefalia. (5 imágenes)
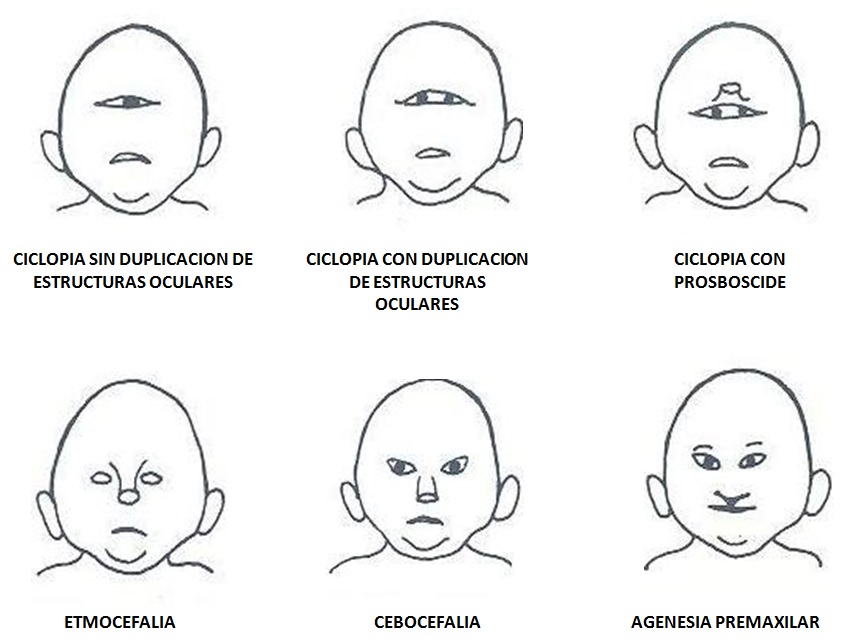
2.1.8.1. Anomalías craneofaciales asociadas a holoprosencefalia
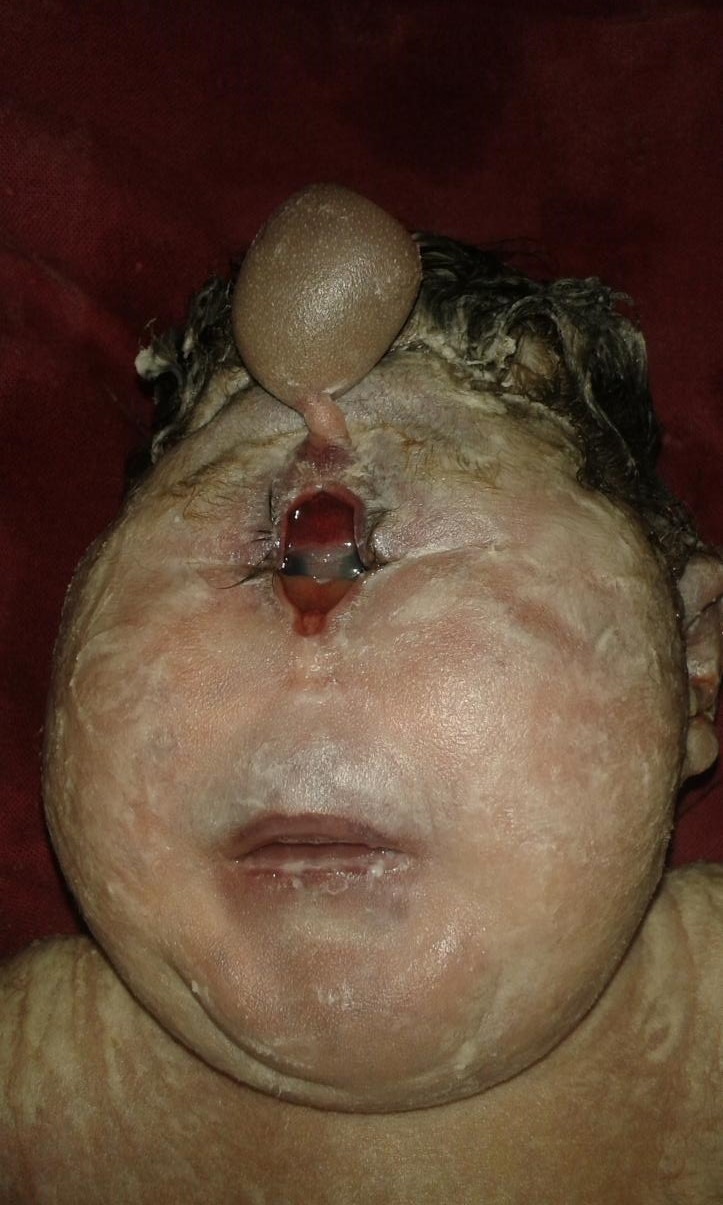
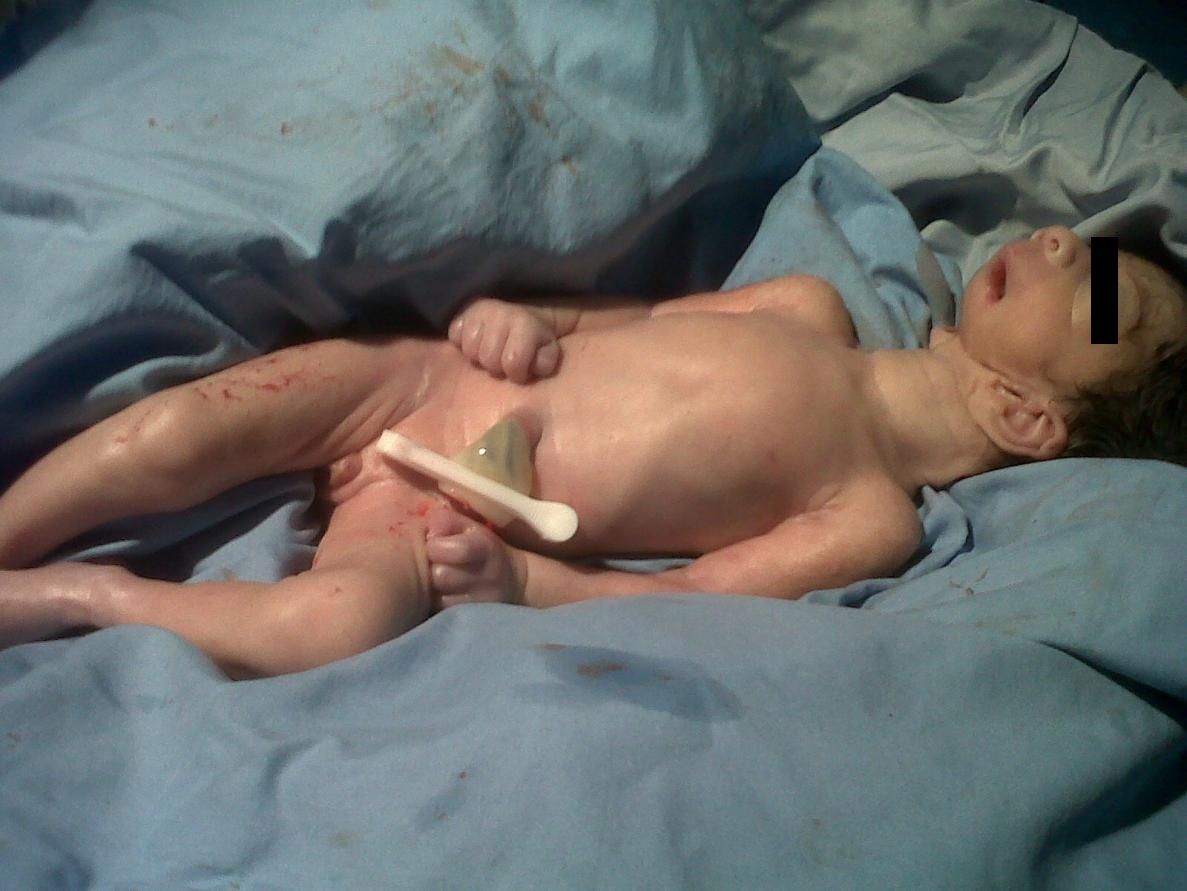
- Ciclopía: Corresponde al subtipo más severo dentro de este espectro. Consiste en una única órbita ubicada en línea media. Usualmente presenta una probóscide situada sobre la órbita y ausencia de estructuras nasales.
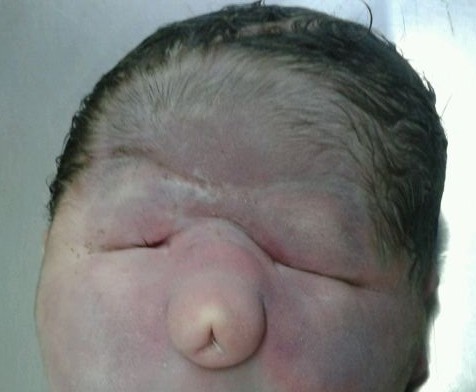
- Etmocefalia: Es el subtipo menos frecuente, se presenta con una prosbóscide entre dos órbitas marcadamente hipotelóricas, usualmente con microftalmía y ausencia de estructuras nasales. Ambas, la etmocefalia y la ciclopía se asocian con holoprosencefalia alobar en la mayoría de los casos.
- Cebocefalia: Presenta una estructura nasal pequeña, chata y con un único orificio nasal situado por debajo de unas órbitas hipotelóricas e hipoplásicas.
- Ausencia de premaxila: Es la forma más leve en este grupo de malformaciones, presenta fisura labial medial (falta la premaxial, el área debajo del tabique nasal), una nariz pequeña y chata e hipotelorismo.
En general existe una gran correlación entre estas anormalidades faciales severas y la presencia de holoprosencefalia.
Figura 2.1.8.1. Anomalías craneofaciales asociadas a holoprosencefalia (1 imágen)