Definición y descripción
Las displasias esqueléticas (osteocondrodisplasias) son un grupo heterogéneo de múltiples trastornos con afectación del tejido conectivo frecuentemente asociados con complicaciones ortopédicas y grados variables de estatura baja y discapacidad. Estos trastornos se diagnostican según criterios radiográficos, clínicos y moleculares. Se sospecha este tipo de cuadros en recién nacidos que presenten talla corta o desproporción entre miembros y tronco o tórax estrecho. Para este tipo de cuadros es fundamental contar con los datos antropométricos, un registro radiográfico del niño (radiografía de cuerpo entero frente y perfil) y fotos de recién nacidos y las radiografías. Algunas displasias requieren evaluar el perfil fosfato-cálcico del afectado.
Etiología e impacto
Hay más de 450 displasias esqueléticas definidas actualmente y alrededor de 100 pueden tener una manifestación clara al momento del nacimiento. Aunque cada displasia esquelética es relativamente rara, colectivamente la incidencia al nacimiento de estos trastornos es casi 1en 5.000. En su gran mayoría son de causa genética mendeliana, es decir, por alteración de un gen principal; esto puede implicar riesgos altos de recurrencia en un futuro embarazo (Ej. Displasia ósea Costillas cortas con polidactilia de herencia autosómico recesiva, tiene un riesgo de recurrencia de un 25%). Hay algunas que tienen alta letalidad perinatal.
Aspecto genético del manejo inicial
Importancia de los hallazgos radiológicos en el proceso diagnóstico: confirma si se trata de una displasia o no, y puede permitir el diagnóstico específico de esa displasia según el tipo de compromiso, por ejemplo:
- Si la desproporción es a predomino del tronco es importante verificar si las vértebras tienen platispondilia (achatamiento).
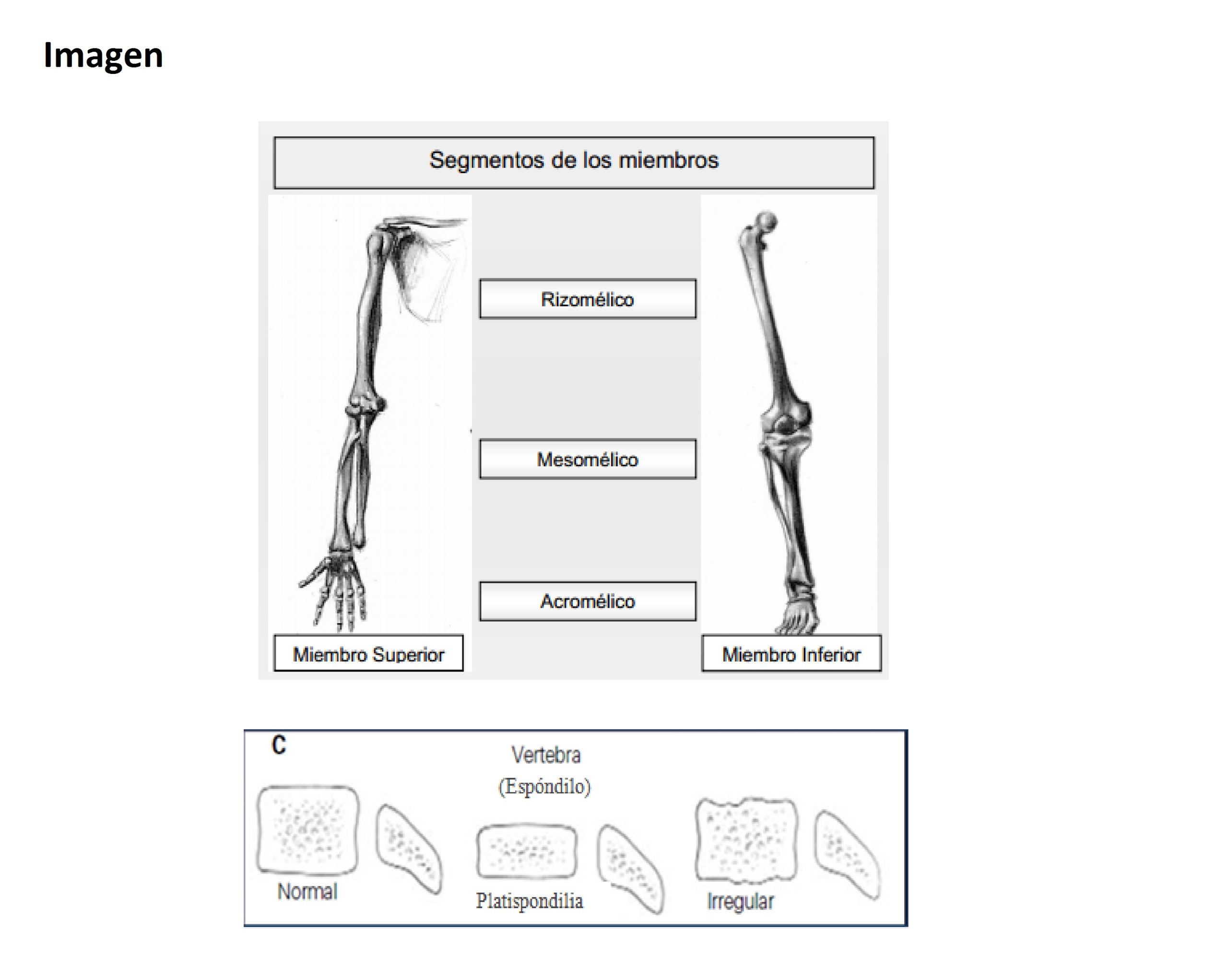
- Si la desproporción es a predominio de miembros, hay que definir si el acortamiento del segmento superior (rizomélico: húmero y fémur), segmento medio (mesomélico: radio, cúbito, tibia y peroné) o segmento distal (acromélico: manos y pies) porque esto puede reducir el diagnóstico Si tiene compromiso en la osificación, tener en cuenta en qué sector, por ejemplo, de los huesos largos: epífisis/metáfisis/diáfisis.
Llegar al diagnóstico especifico permitirá el asesoramiento genético a la familia.
Displasias óseas, algunos ejemplos frecuentes
Acondroplasia:
Afecta a 1/15.000 nacimientos. Se caracteriza por extremidades cortas (acortamiento mayor de la parte proximal de miembros superiores, llamado acortamiento rizomélico), hiperlordosis, manos pequeñas (en tridente) y macrocefalia con frente alta y nariz en silla de montar. Radiológicamente se observa:
- Macrocefalia con base del cráneo y foramen magnum pequeños
- Disminución de la distancia interpeduncular en sentido céfalo-caudal en vértebras lumbares (rx frente), este espacio normalmente debe ensancharse en sentido céfalo-caudal.
- Pedículos cortos (en radiografías de perfil).
- Huesos iliacos redondeados y planos, acetábulo horizontalizado y espina sacrociática pequeña
El desarrollo mental es normal. El diagnóstico se basa en los resultados radiológicos. La enfermedad es autosómica dominante, aunque alrededor del 80% de los pacientes afectados son hijos de padres no-afectados. Estos casos se deben a mutaciones nuevas, que pueden correlacionarse en algunos casos con la edad paterna avanzada como factor de riesgo.
Displasia tanatofórica
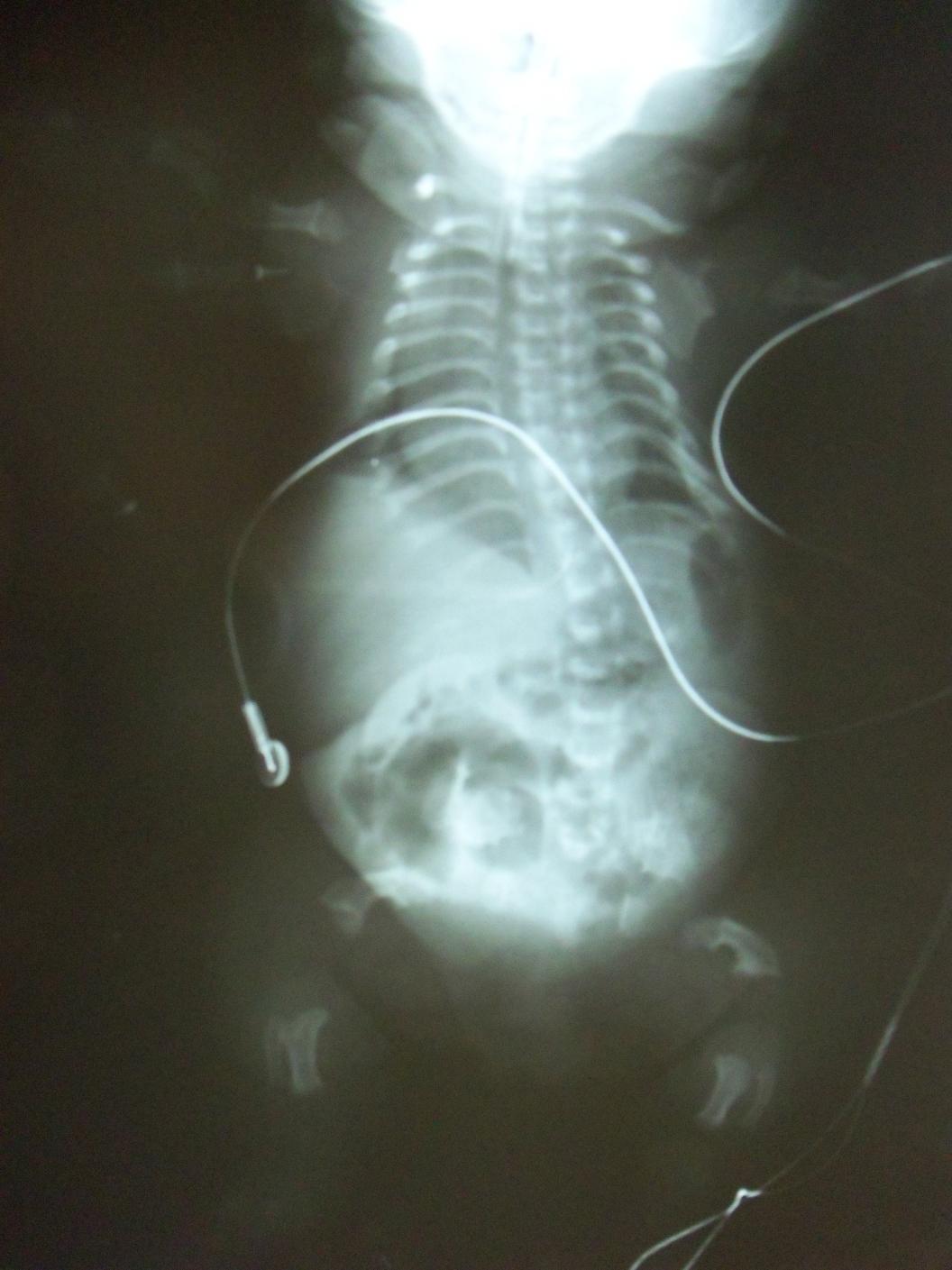
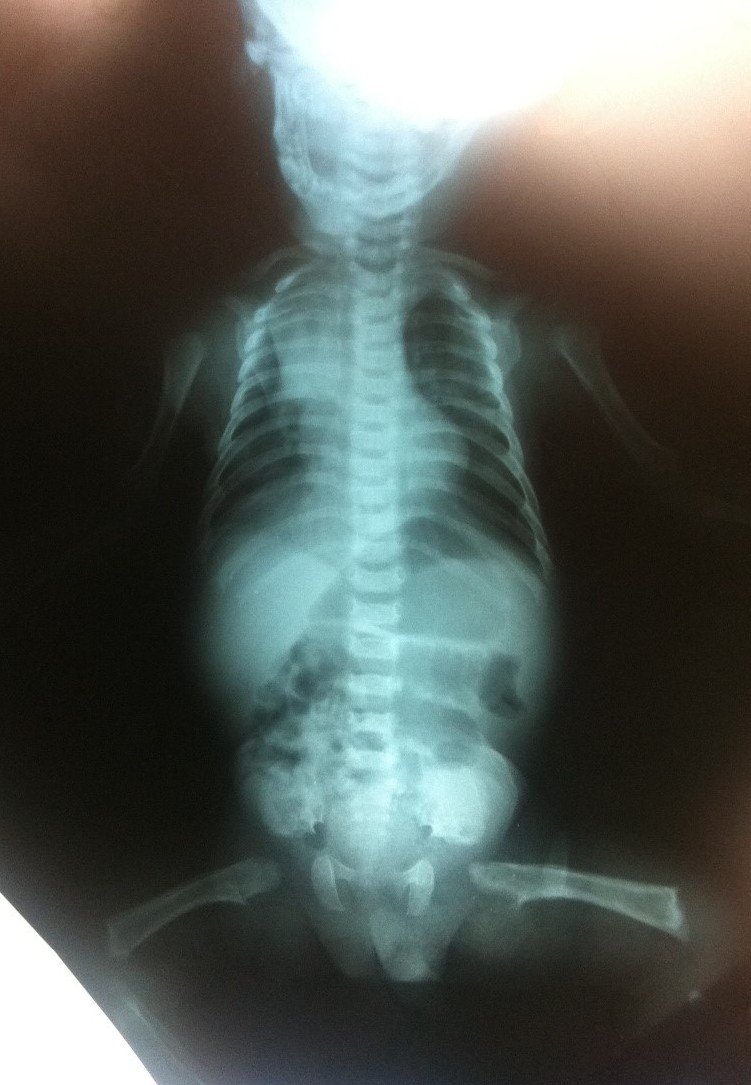
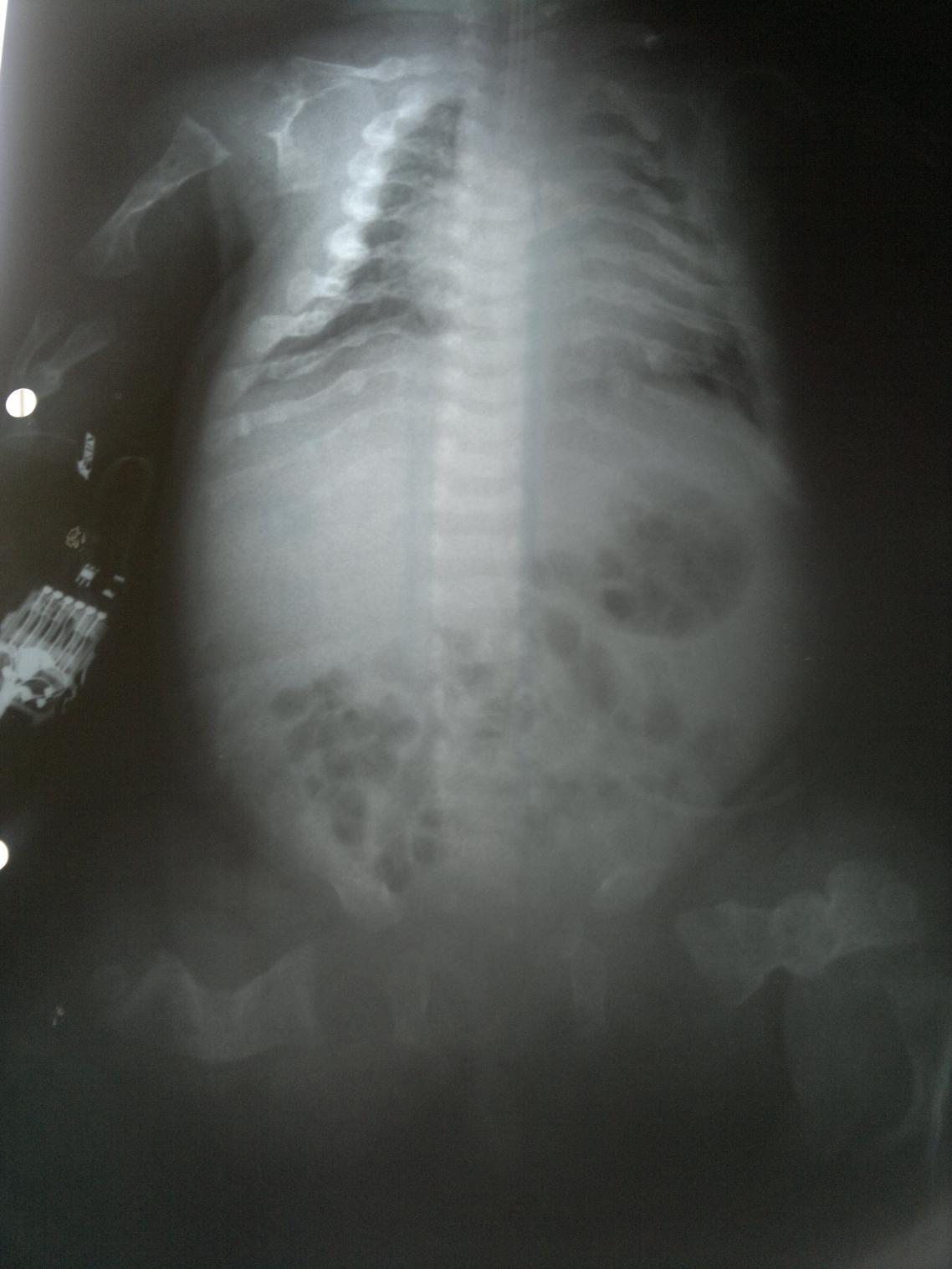
Es la displasia esquelética letal más común. Se reconocen dos variantes, tipos I y II. Ambos subtipos se consideran displasias esqueléticas letales; la mayoría de los bebés afectados mueren por insuficiencia respiratoria en las primeras horas o días de vida. Este cuadro se caracteriza por un acortamiento severo de las extremidades, un tórax estrecho en campana, macrocefalia, fontanela anterior amplia, puente nasal bajo y una longitud del tronco relativamente normal, vértebras achatadas (platispondilia). En algunos casos se asocia a una craneosinostosis severa (cráneo en trébol) así como anomalías neurológicas (hidrocefalia, trastornos de migración neuronal, entre otros). La displasia tanatofórica es causada por una mutación de novo. El riesgo de recurrencia es bajo.
Osteogénesis imperfecta
Es una afección en la que ocurren fracturas con traumas leves o ausentes y es causada por un defecto genético que afecta el colágeno tipo 1. Existen al menos 9 tipos diferentes de osteogénesis imperfecta (OI), de los cuales los tipos II y III tienen mayores manifestaciones clínicas en el neonato.
Osteogénesis imperfecta Tipo II
Es la forma más severa de OI. Los niños que nacen con este tipo de OI presentan fracturas perinatales, miembros poco desarrollados y curvos, y tienen los huesos extremadamente frágiles. Con frecuencia fallecen poco después de nacer (una de las razones de esta mortalidad temprana son las hemorragias internas que se producen como consecuencia de las numerosas fracturas). El diagnóstico prenatal es posible: en la ecografía se puede apreciar la curvatura de los miembros y determinadas fracturas. En las radiografías se pueden ver fracturas múltiples de costillas, mineralización mínima de la calota, compresión marcada de huesos largos, vertebras aplanadas y fracturas en diferentes tiempos de evolución. Las escleróticas de estos pacientes suelen verse azuladas. El cuadro resulta frecuentemente de una mutación de novo por lo cual el riesgo de recurrencia en hermanos es bajo. Sin embargo, en 5% de los casos puede repetirse lo cual se explica por la presencia de un mosaicismo germinal en alguno de los padres.
Osteogénesis imperfecta Tipo III o progresiva deformante
El pronóstico de la osteogénesis imperfecta tipo III es severo. Los niños afectados sufren con frecuencia fracturas espontáneas. El diagnóstico prenatal es posible mediante ecografía.. En las radiografías pueden verse: platispondilia de los cuerpos vertebrales, costillas finas, múltiples fracturas y epífisis comprometidas (se puede observar el “signo del pochoclo”). Las escleróticas de estos pacientes también suelen verse azuladas.
huesos largos delgados con chas
El siguiente es un cuadro comparativo entre las OI tipo I, II y III
| Tipo | Herencia | Severidad | Hallazgos radiográficos | Deformidad ósea | Talla | Afectación
Dental |
Escleróticas |
| I | Autosómica Dominante | Leve | Desde pocas a 100 fracturas | Infrecuente | Normal o levemente disminuida | Rara | Azules |
| II | Mutación de novo | Letal perinatal | Múltiples fracturas costales, mínima mineralización de la calota, platispondilia, marcada compresión de huesos largos | Severa | Talla baja severa | Sí | Azules |
| III | Autosómica Dominante | Severa | Costillas finas, plastispondilia, huesos finos con fracturas | Moderada a severa | Talla baja | Sí | Azules |
Para las familias de los pacientes con es importante contar con una consulta con un servicio de genética. Si bien las OI tipo II y III ocurren por mutaciones de novo (autosómica dominante) y por ende el riesgo de recurrencia bajo, existen casos de mosaicismo gonadal en los padres que explican que el riesgo de recurrencia sea de un 5%.
Figura 2.11. Displasias óseas. (4 imágenes)